Sept 2021 Edition

Genetic Diseases of the Kidney
This quarter’s eDigest starts with a selection of papers from Kidney International addressing genetic kidney conditions.
More than 60 genetic diseases, ranging from relatively common conditions to extremely rare syndromes, are currently known to affect the kidneys.
Recent developments in genetics, such as next-generation sequencing, have improved the diagnostic potential for patients with chronic kidney disease and offer significant potential for precision medicine in the field of kidney transplantation. Developing the best way to implement these methods in clinical practice presents an ongoing challenge in kidney care.
Special expertise is needed to diagnose and treat these conditions. The ISN Journals are dedicated to presenting the latest clinical investigations and debates to help address the special issues experienced by those with genetically-determined kidney diseases.
KIDNEY INTERNATIONAL ARTICLES |
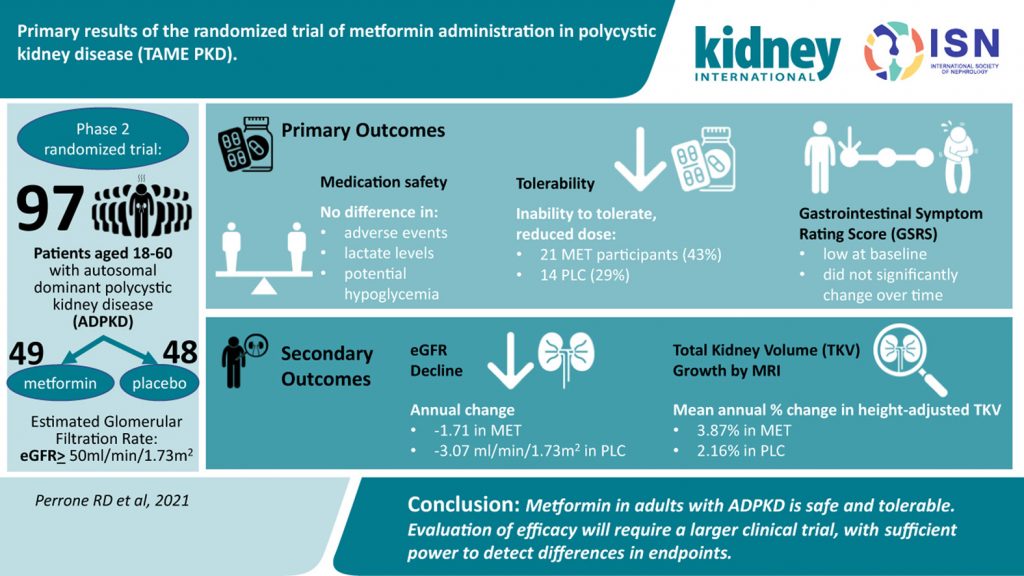
Primary Results of the Randomized Trial Method of Metformin Administration in Polycystic Kidney Disease (TAME PKD)
Autosomal dominant polycystic kidney disease (ADPKD) is characterized by the growth of kidney cysts and a decline in glomerular filtration rate (GFR).
A phase 2, two-year trial builds on studies demonstrating that Metformin was found to impact cystogenesis in preclinical models of polycystic disease and may be a promising candidate for clinical investigation in ADPKD.
Primary outcomes of this randomized trial suggest that Metformin use in adults with ADPKD was safe and tolerable while not reducing estimated GFR decline to a significant degree. The authors suggest the need for a larger trial to detect differences in endpoints and evaluate treatment efficacy.
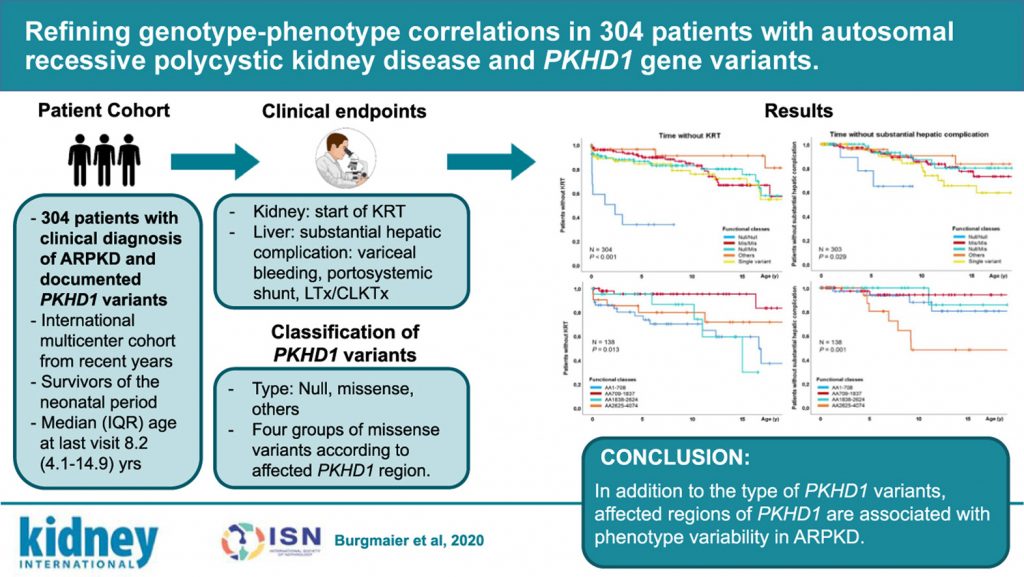
Refining Genotype-phenotype Correlations in 304 Patients with Autosomal Recessive Polycystic Kidney Disease and PKHD1 Gene Variants
Autosomal recessive polycystic kidney disease (ARPKD) is a severe disease of early childhood that is clinically characterized by fibrocystic changes of the kidneys and the liver.
This observational study analyzed a deep clinical dataset of 304 patients with ARPKD from two independent cohorts and identified novel genotype-phenotype correlations during childhood and adolescence.
The results improve understanding of genotype-phenotype correlations in pediatric ARPKD patients and lay the foundation for more precise and personalized counseling and treatment approaches.
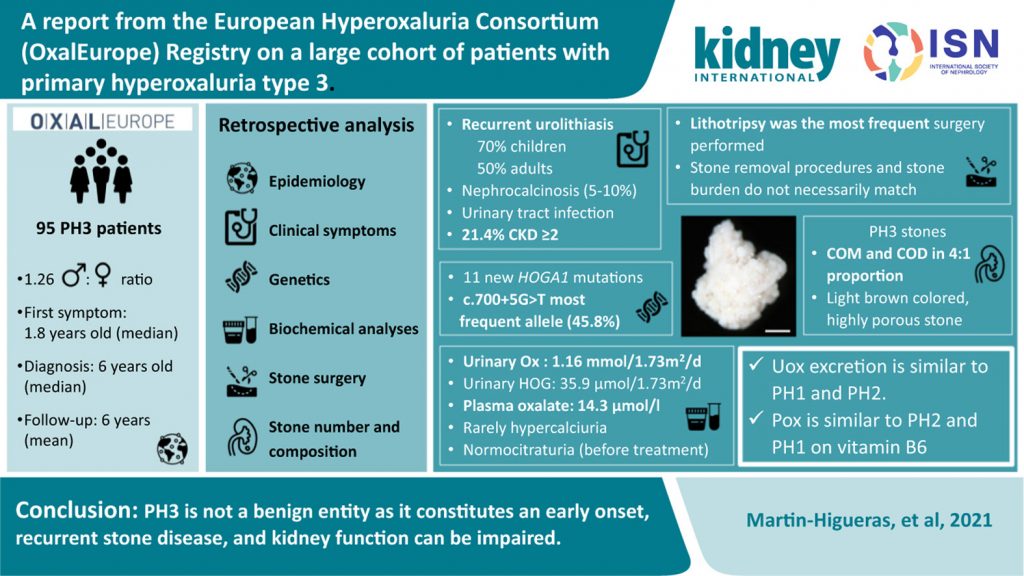
A report from the European Hyperoxaluria Consortium (OxalEurope) Registry on a large cohort of patients with primary hyperoxaluria type 3
The scarcity of outcome data in primary hyperoxaluria type 3 (PH3), a less severe form of the primary hyperoxalurias (PHs) with a low risk of chronic kidney disease, prompted the authors to conduct a retrospective analysis of the largest PH3 cohort to date.
The results demonstrated that urolithiasis was the major clinical feature observed in 70% of pediatric and 50% of adult patients. Alongside other results, the investigation shows that PH3 is more comparable to PH1 and PH2 than smaller studies would suggest. It is the most favorable PH type but can lead to early-onset, recurrent urinary stone disease, and impaired kidney function.
Establishing a Nephrology Genetic Clinic
Recent advances in clinical molecular screening have created the potential for a genetics revolution within nephrology practice.
Genetic testing can provide a firm diagnosis and the genetic information to change management, treatment options, and eligibility for clinical trials, as well as guide transplantation decisions and allow patients to make informed choices.
This editorial discusses the challenges of implementing genetic testing for kidney health diagnostics and the experience of addressing them successfully in a tertiary medical center.
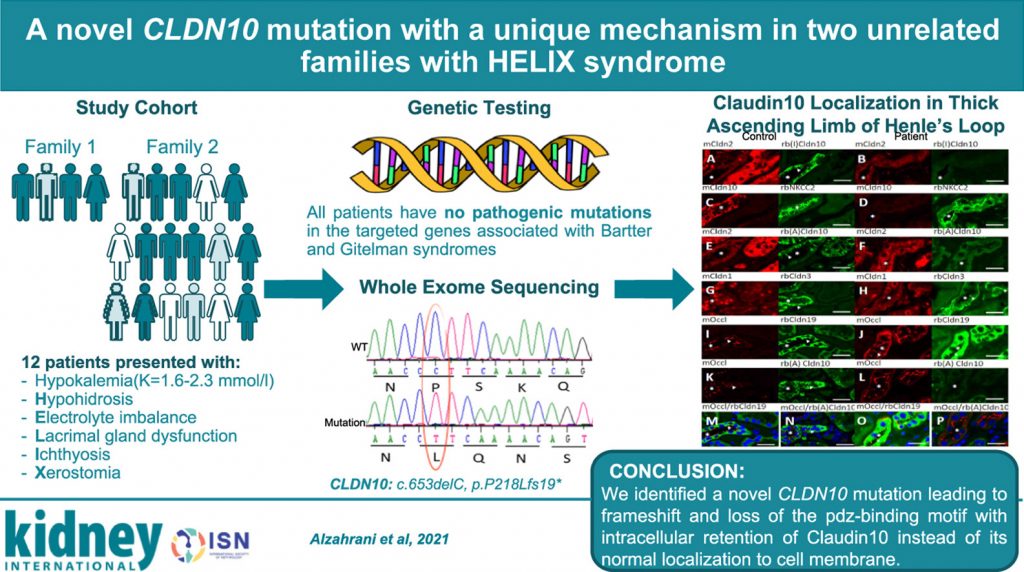
A novel claudin-10 mutation with a unique mechanism in two unrelated families with HELIX syndrome
This investigation describes two unrelated Saudi families with HELIX syndrome due to a novel CLDN10 mutation with a unique mechanism of CLDN10 inactivation.
The two families include 12 affected individuals who presented with hypokalemia and other features of HELIX syndrome, notably hypohidrosis, electrolyte imbalance, lacrimal gland dysfunction, ichthyosis, and xerostomia due to claudin-10 (CLDN10) mutations.
The underlying mutation was detected by whole-exome sequencing, confirmed by Sanger sequencing, and functionally indicated by RT-PCR, electrophysiological studies, and immunohistochemical staining of transfected HEK293 and MDCK C7 cells, and skin and kidney biopsy tissues.
The analysis demonstrates that the novel CLDN10 mutation identified in the two families disrupted the C-terminus pdz-binding motif of claudin-10, causing HELIX syndrome.
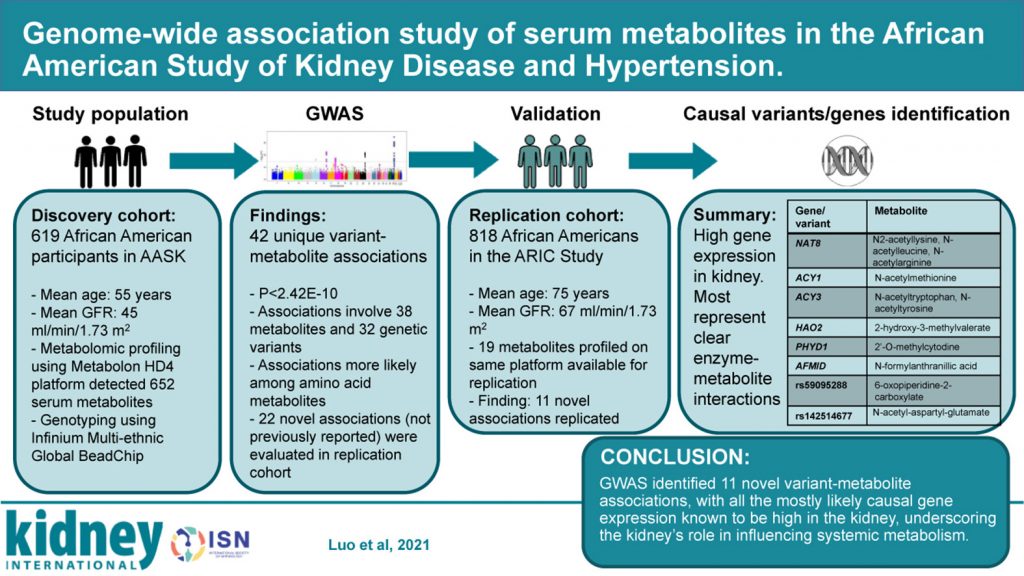
Genome-wide association study of serum metabolites in the African American Study of Kidney Disease and Hypertension
The genome-wide association study (GWAS) offers a powerful way to study genetic determinants of disease traits and generate insights into disease pathophysiology.
To explore the hypothesis that novel genetic-metabolite associations may be identified in a unique population of African Americans with a lower glomerular filtration rate (GFR), the authors conducted a GWAS of 652 serum metabolites in 619 participants in the African American Study of Kidney Disease and Hypertension, a clinical trial of blood pressure lowering and antihypertensive medication in African Americans with chronic kidney disease.
The results demonstrate that extensive metabolite profiling in an African American population with chronic kidney disease helped identify novel genome-wide metabolite associations, providing clues about substrate specificity and the key roles of enzymes in modulating systemic levels of metabolites.
APOL1 Genotyping in Kidney Transplantation: To Do or Not to Do, That Is the Question? (Pro)
While acknowledging that genetic testing has risks, that it is a personal choice, and should never be undertaken lightly, the authors present evidence supporting APOL1 genotyping in kidney transplantation.
They outline the usefulness of the National Institutes of Health-sponsored “APOL1 Long-Term Kidney Transplantation Outcomes” (APOLLO) Network to provide critical information on the safety of live kidney donation and transplantation outcomes from APOL1 high-risk donors.
APOL1 Genotyping In Kidney Transplantation: To Do or Not to Do, That Is the Question? (Contra)
The authors outline the case against routine APOL1 testing in kidney transplantation in the hope of encouraging a more balanced approach to the care of donors and recipients alike. They question the rationale for routine testing of all African American donors and recipients for 2 APOL1 renal risk variants when 87% of African Americans do not carry 2 APOL1 renal risk variants.
They suggest that such strategies, though well-intentioned, may eliminate patient autonomy in decision-making and even exacerbate existing disparities.
APOL1 Genotyping in Kidney Transplantation: A Look into the Future
The author presents an overview of APOL1 kidney risk variants and the pro and contra arguments presented in the Kidney International “controversies” articles above. He urges priority for developing prediction models that can identify living donors at risk of developing CKD and deceased donor kidneys at risk of allograft failure in the presence of APOL1 KRVs with better precision.
Critical data from APOLLO will improve discussions between health care providers, patients, and potential kidney donors in the informed clinical decision-making process.
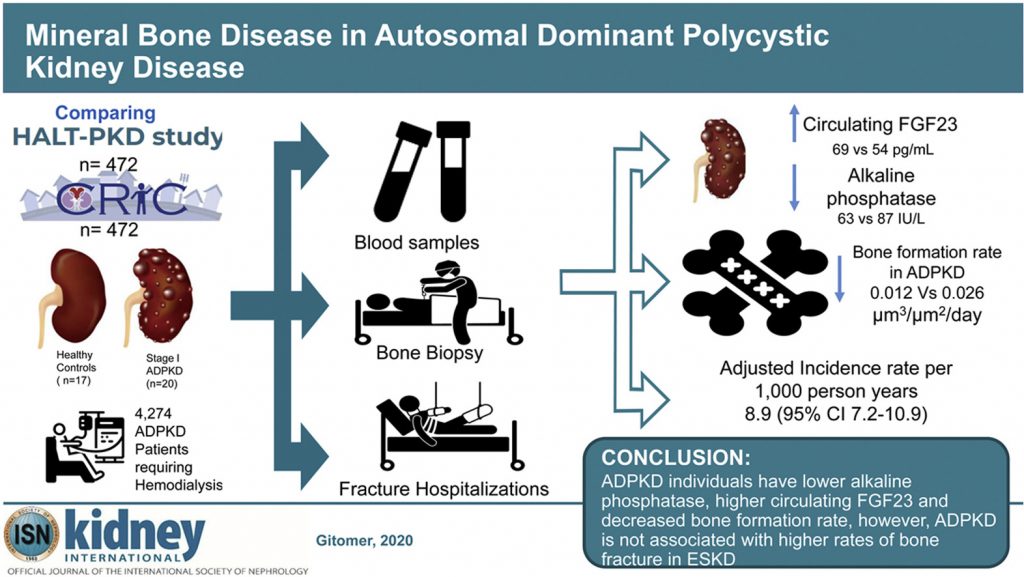
Mineral Bone Disease in Autosomal Dominant Polycystic Kidney Disease
Mice with disruption of Pkd1 in osteoblasts demonstrate reduced bone mineral density, trabecular bone volume, and cortical thickness.
The bone phenotype in adult patients with autosomal dominant polycystic kidney disease (ADPKD) with stage I and II chronic kidney disease has not yet been investigated. To address this gap, the authors characterized biochemical markers of mineral metabolism, examined bone turnover and biology, and estimated the fracture risk in a sample of 944 patients with ADPKD and other causes of kidney disease.
The results indicate that individuals with ADPKD have lower alkaline phosphatase, higher circulating intact fibroblast growth factor 23, and decreased bone formation rate. ADPKD is not, however, associated with higher rates of bone fracture in ESKD.
KIDNEY INTERNATIONAL REPORTS ARTICLE |
Numerous articles in the September issue of KI Reports reveal disease patterns and treatment advances for patients with nephrotic syndrome (NS) and its related conditions.
Timing of COVID-19 Vaccine in the Setting of Anti-CD20 Therapy: A Primer for NephrologistsRepository Corticotropin Injections Promote Clinical Remission and TREG Expansion in Membranous Nephropathy
Since membranous nephropathy (MN) is a known cause of nephrotic syndrome (NS), treatment may help prevent NS progression. In a small group of patients with MN, repository corticotropin injection on T- and B-cell subsets (including TREG) was effective in remission after one year of treatment. Acthar Gel increased TREG induction, which could be the reason for its therapeutic effects.
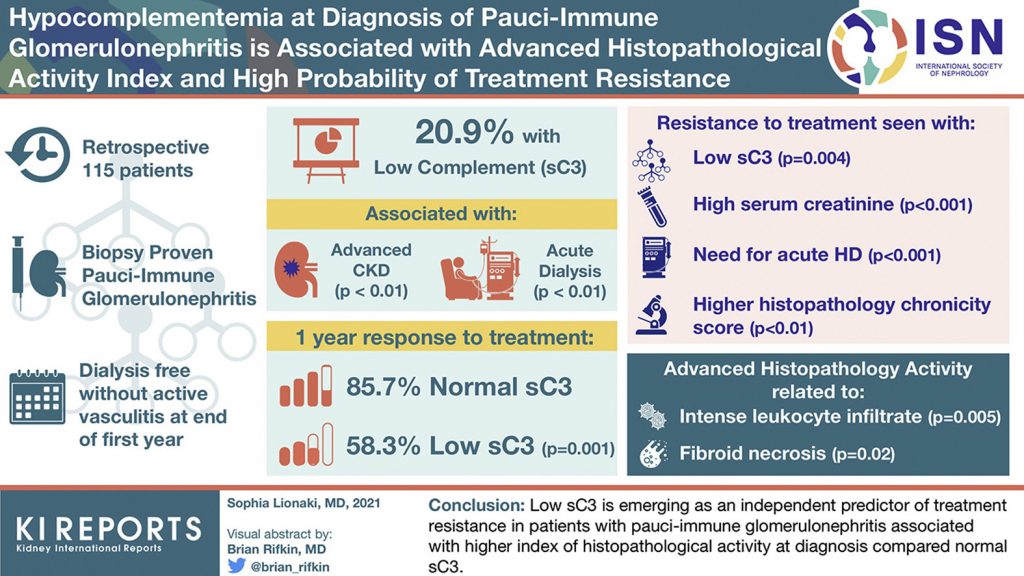
Hypocomplementemia at Diagnosis of Pauci-Immune Glomerulonephritis is Associated with Advanced Histopathological Activity Index and High Probability of Treatment Resistance
Serum complement C3 (sC3) is emerging as an independent predictor of treatment resistance in patients with pauci-immune glomerulonephritis (PIGN). Researchers in Greece found that low sC3 was associated with more disease activity at diagnosis and treatment resistance in patients with PIGN.
Timing of COVID-19 Vaccine in the Setting of Anti-CD20 Therapy: A Primer for NephrologistsClinical Outcomes of People With Fabry Disease — ANZDATA Registry Study
Clinical nephrologists in Australia analyzed data from The Australian and New Zealand Dialysis and Transplant (ANZDATA) registry to provide the first in-depth report on clinical outcomes of patients with Fabry Disease (FD) receiving renal replacement therapy (RRT) in Australia and New Zealand. The major cause of death among those with FD on dialysis was cardiovascular disease. In both transplant and post-transplant recipients, FD was associated with increased mortality.